
The OBP OfficeSPEC® Disposable Side-Opening Speculum is indicated for use on women undergoing a procedure requiring vaginal access and exposure (e.g., for a pelvic exam or Pap smear). The device uses opposing bi-valve plastic blades to separate the vaginal wall and is intended to expose the interior of the vagina and exterior of the cervix. It may be used with or without a removable light source.
Lighted Specula Kits and Accessories
From

Product codes
| Product | Order Code |
|---|---|
| OfficePack for Hysteroscopy (1 Box of 5) | C040100 |
| Fluid Management Pack (1 Box of 5) | C050100 |
Get In Touch With Us
We'd love to hear from you. How can we help?
Brochures, Catalogs & Flyers
Product Brochures
Product Brochures
Product Catalogs
Product Catalogs
Related Products

Wallach® Foot Pedal-Operated Pencils
Sterile, single-use only. For use with Quantum 2000® and other comparable electrosurgical generators. Reusable adapter required. Use with any standard...

Wallach® Hand Switch-Operated Pencils
Sterile, single-use only. For use with Quantum 2000®and other compatible electrosurgical generators. Convenient CUT and COAG push buttons.

Wallach® LL100™ System (N2O)
The LL100™ is a cryosurgical unit used for ablative-type surgical technique.
Webinars
Articles
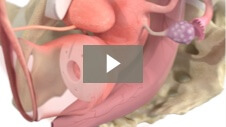
Milex Shaatz Pessary
Animation demonstrating insertion of Shaatz Pessary for 1st and 2nd degree pelvic organ prolapse.
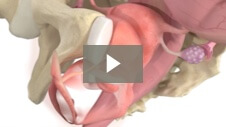
Milex Hodge Pessary
Animation demonstrating insertion of Hodge Pessary for Stress Urinary Incontinence.
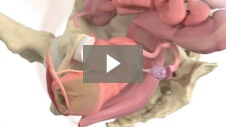
Milex Inflatoball Pessary
Animation demonstrating insertion of Inflatoball Pessary for 2nd and 3rd degree pelvic organ prolapse/procidentia.
Support & Compliance
Our global team is committed to providing the highest standards of service and support.

Batch Certificates
Use this tool to enter your batch number and download the corresponding certificate of analysis.

Service
We offer a range of contract options to suit your needs: preventative maintenance and service, reliable access to spare parts, product training, and online handling of service requests.

Get In Touch With Us
We'd love to hear from you. How can we help?